DVP-IO x SOPA/Cellpose#
In this tutorial, we will run the cell segmentation pipeline implemented in sopa [BMG+24] to perform cell segmentation of a multi-channel fluorescence image in the .czi format with cellpose. We will use dvp-io to read the image from its native format into the sopa-supported spatialdata format. Subsequently, we will run the sopa cell segmentation pipeline. Finally we will use dvp-io to export the shapes as LMD-compatible .xml file.
Load modules#
This tutorial requires you to have sopa with cellpose support installed in your local environment.
conda create -n dvpio python=3.11 -y && conda activate dvpio
pip install dvp-io
pip install "sopa[cellpose]"
import sopa
from dvpio.read.image import read_czi
from dvpio.write.shapes import write_lmd
import spatialdata_plot # noqa
import spatialdata as sd
import numpy as np
import matplotlib.pyplot as plt
Load data#
We will use a sample .czi image (3 channels) kindly provided by Sophia Mädler from the scPortrait package [SMW+23] and convert it to a spatialdata object using dvpio.read.image.read_czi. Note that the converted spatialdata object must be written to disk for the rest of the notebook to function. This is because some features of read_czi rely on C modules that cannot be parallelized by Dask, which sopa’s parallelization backend depends on.
## Uncomment to obtain data
# ! wget -O "./data/example.czi" "https://datashare.biochem.mpg.de/s/4jH87DiwvxoM4Qd/download"
sdata = sd.SpatialData()
# Get all 3 channels from czi image
sdata.images["czi"] = read_czi("./data/example.czi", channels=[0, 1, 2])
# Write sdata to disk
sdata.write("./data/scportrait-czi.sdata.zarr")
INFO The Zarr backing store has been changed from None the new file path: data/scportrait-czi.sdata.zarr
# Re-read sdata object from disk
sdata = sd.read_zarr("./data/scportrait-czi.sdata.zarr")
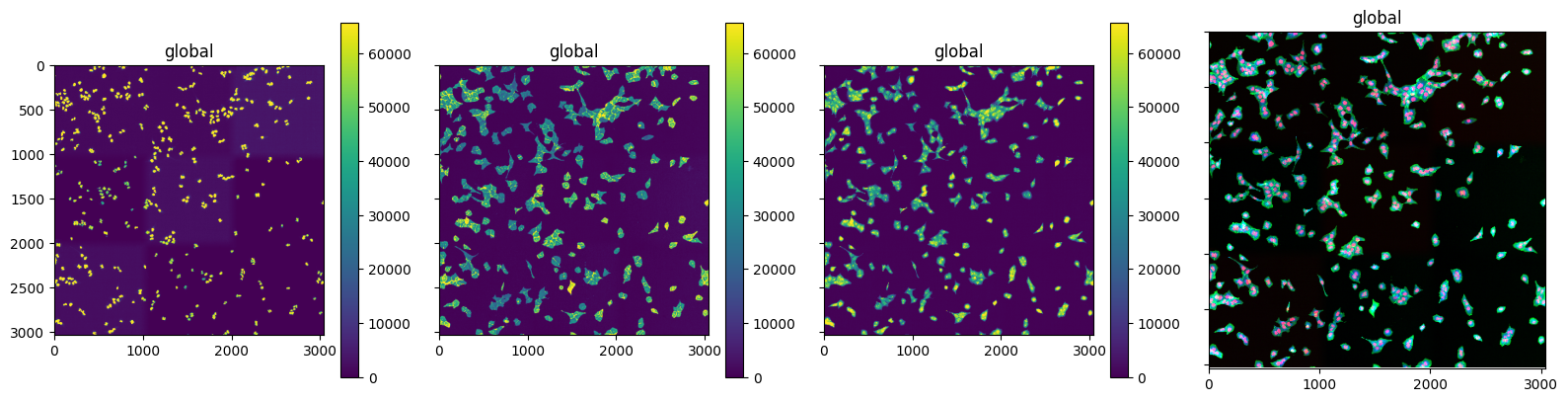
We can plot the 3 channels. Channel 0 corresponds to the nuclei channel and 1 to the cytosol channel
fig, axs = plt.subplots(1, 4, figsize=(16, 4), sharex=True, sharey=True)
for idx in range(3):
sdata.pl.render_images("czi", channel=idx).pl.show(ax=axs[idx])
sdata.pl.render_images("czi").pl.show(ax=axs[3])
plt.tight_layout()
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
Run sopa#
We can now run sopa. Sopa first creates smaller image patches that can be passed to cellpose, using the sopa.make_image_patches method. Subsquently, we can run the cellpose wrapper sopa.segmentation.cellpose. Note that you can also pass your own pretrained models to via the pretrained_model="/path/to/pretrained/model" argument. Here, we are using the default cyto3 model.
sopa.make_image_patches(sdata, patch_width=500, patch_overlap=50, image_key="czi")
# Set dask as parallelization backend
sopa.settings.parallelization_backend = "dask"
sopa.segmentation.cellpose(sdata, channels=[1, 0], diameter=50, model_type="cyto3", image_key="czi")
# Runs in ca. 30s on MacOS/M3
[ ] | 0% Completed | 0.4s
[########################################] | 100% Completed | 23.3s
We can inspect the cell segmentation in a smaller patch and we can see that the model performed well on this dataset.
(
sdata.query.bounding_box(
axes=("x", "y"),
min_coordinate=np.array([0, 0]),
max_coordinate=np.array([600, 600]),
target_coordinate_system="global",
)
.pl.render_images("czi")
.pl.render_shapes("cellpose_boundaries", fill_alpha=0, outline_alpha=1)
.pl.show()
)

Write shapes#
In many cases, we are now interested in exporting the obtained shapes to a Leica Microdissection Microscope. We can use the dvpio.write.write_lmd function to export shapes in a compatible format. The function wraps the py-LMD package [SMW+23].
The function also relies on adding calibration points
sdata
SpatialData object, with associated Zarr store: /Users/lucas-diedrich/Documents/Projects/scverse/spatialdata/spatialdata-io/dvp-io/dvp-io/docs/tutorials/data/scportrait-czi.sdata.zarr
├── Images
│ └── 'czi': DataArray[cyx] (3, 3038, 3040)
└── Shapes
├── 'cellpose_boundaries': GeoDataFrame shape: (579, 1) (2D shapes)
└── 'image_patches': GeoDataFrame shape: (49, 3) (2D shapes)
with coordinate systems:
▸ 'global', with elements:
czi (Images), cellpose_boundaries (Shapes), image_patches (Shapes)
cell_shapes = sdata.shapes["cellpose_boundaries"]
We will also need to select calibration points in the image.
## Uncomment to select calibration points interactively
# interactive = Interactive(sdata)
# interactive.run()

## Uncomment to select interactively selected calibration points
# calibration_points = sdata.points["calibration_points_image"]
# Use these points for reproducibility
calibration_points = sd.models.PointsModel.parse(
np.array([[251.03580019, 151.55537058], [1999.99218284, 36.1721564], [109.75405585, 2953.58610685]])
)
Lastly, we need to define an affine transformation that is used to translate the image coordinates into LMD coordinates.
affine_transformation = np.array([[0, -10, 0], [10, 0, 0], [0, 0, 1]])
We can use this information to write an LMD-compatible xml file that contains the segmented cells as shapes
write_lmd("./data/lmd.xml", cell_shapes, calibration_points, affine_transformation)
[0. 0.]
[ 0. -10000.]
[10000. 0.]