Real-world example - Analysis of scDVP liver zonation dataset#
In this example we showcase how spatialdata can be leveraged in the analysis of real-world DVP data. For this purpose we utilize a small subset of the previously published dataset by [RTS+23] (Rosenberger et al, 2023), specifically the sample m1a. This datasets includes
IF images (as
.tiff) of the mouse liver, including the markers Glutamine Synthethase, E-Cadherin, cell membrane marker (WGA), and phalloidin (cytoskeleton)Cell segmentation shapes of the profiled cells as LMD-compatible
.xmlfileProteomics measurements of 77 single shapes as diann protein groups report (
.tsv)
We will see how we can use spatialdata to jointly store and visualize these different modalities.
Packages#
To process the different modalities, we import the respective readers from dvpio (see the FAQ) for a full list of available readers). Additionally, we will import some scientific software packages, including spatialdata
import numpy as np
import pandas as pd
import matplotlib as mpl
import matplotlib.pyplot as plt
import spatialdata as sd
import spatialdata_plot # noqa
from dvpio.read.image import read_custom
from dvpio.read.omics import parse_df
from dvpio.read.shapes import read_lmd
Get data#
# %%capture
# %%bash
# downloadpath="./scdvp"
# mkdir -p $downloadpath
# wget -O $downloadpath/scdvp-rosenberger2023.zip "https://datashare.biochem.mpg.de/s/Z0BBq65fFHX3ai3/download"
# unzip -d $downloadpath $downloadpath/scdvp-rosenberger2023.zip
# rm $downloadpath/scdvp-rosenberger2023.zip
image_path = "./scdvp/images"
lmd_path = "./scdvp/shapes/EXP-220815_m1A_plate-2.pylmd.xml"
protein_group_df_path = "./scdvp/omics/PXD038699_proteintable.tsv"
Create spatialdata object#
We will successively add attributes to our spatialdata object. Let’s initialize an empty object:
sdata = sd.SpatialData()
Load images#
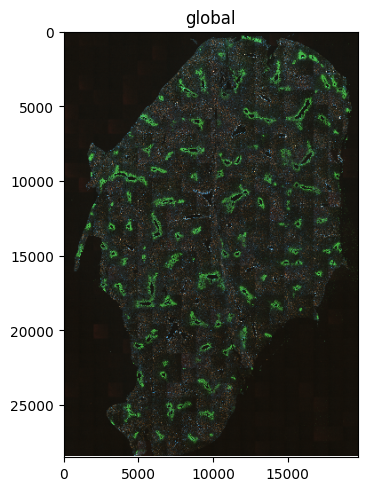
First, we will add the tiff images using the respective dvpio function and visualize it
sdata.images["m1a_image"] = read_custom(path=f"{image_path}/S-BIAD596_m1A_*.tif")
INFO no axes information specified in the object, setting `dims` to: ('c', 'y', 'x')
norm = mpl.colors.Normalize(vmin=0, vmax=4000)
sdata.pl.render_images("m1a_image", norm=norm).pl.show()
INFO Rasterizing image for faster rendering.
Load shapes#
Next, we load the shapes. To do this, we will first need to define the calibration points in the image. As the calibration points are not visible in the image, we pass the values that were used in the original publication (link). Alternatively, you can also interactively select the points with napari_spatialdata, see Tutorial 2
# Calibration points were determined at 0.135545805x resolution in a coordinate system that was centered at the image center
# + we need to scale them to 1x resolution (x1/0.135545805)
# + shift the coordinate system to the upper left corner of the image (+image width/2, image width/2)
url = "https://raw.githubusercontent.com/MannLabs/single-cell-DVP/cf9d588849f84409c2e0ce6e3703cc69a1823503/data/meta/coordinates_meta.txt"
df = pd.read_csv(url, sep="\t")
df["X_scaled"] = df["X"] / df["resolution"]
df["Y_scaled"] = df["Y"] / df["resolution"]
df["X_transformed"] = df["X_scaled"] + sdata.images["m1a_image"].shape[2] / 2
df["Y_transformed"] = df["Y_scaled"] + sdata.images["m1a_image"].shape[1] / 2
calibration_points_image = df.loc[df["Slide"] == "m1A", ["X_transformed", "Y_transformed"]].values
sdata.points["calibration_points_image"] = sd.models.PointsModel.parse(calibration_points_image)
calibration_points_image
array([[ 1836.11676551, 9121.12330304],
[18291.61238192, 3189.23998138],
[ 7121.62375704, 25625.42677448]])
Having defined the calibration points, we can now read the actual shapes from the LMD-compatible .xml file and add them to spatialdata.
sdata.shapes["cell_segmentation"] = (
read_lmd(
lmd_path,
calibration_points_image=sdata.points["calibration_points_image"],
transformation_type="similarity",
)
.set_index("name", drop=False)
.rename_axis(None, axis=0)
)
You should always check/validate whether cell segmentation and image are correctly overlaid. To do so, we select a small subset of the image and take a look at both cell membrane staining (channel 1) and cell segmentation
fig, ax = plt.subplots(1, 1, figsize=(8, 5))
min_coordinate = np.array([6200, 6600])
max_coordinate = min_coordinate + 700
(
sdata.query.bounding_box(
axes=("x", "y"),
min_coordinate=min_coordinate,
max_coordinate=max_coordinate,
target_coordinate_system="global",
)
.pl.render_images("m1a_image", channel=1)
.pl.render_shapes(fill_alpha=0.0, outline_alpha=1, outline_color="#efefef")
.pl.show(coordinate_systems="global", ax=ax)
)
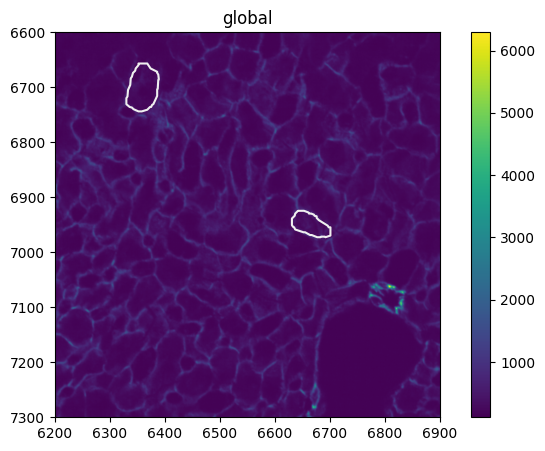
The overlay between shapes and cell membrane stain looks reasonable.
Omics data#
Finally, we add the omics data. In this case, we start from the protein.group dataframe as reported by DIANN (source) and use the :func:dvpio.read.omics.parse_df function to parse the data into a spatialdata-compatible anndata.AnnData container.
Note, that the function expects a processed version of the dataframe: Cell-level metadata must be stored in the dataframe row index and will be stored in the
anndata.AnnData.obsattribute. Gene-level metadata must be stored in the dataframe columns index and will be stored in theanndata.AnnData.varattribute. All values of the dataframe will be stored inanndata.AnnData.X.
# Read DIANN Report
df = pd.read_csv(protein_group_df_path, sep="\t", index_col="protein").T.reset_index(names="run")
# Parse metadata run into individual metadata columns
metadata_columns = [
"date",
"ms",
"A",
"B",
"C",
"method",
"sample_id",
"uniprot_ref",
"aid",
"cell_id",
"D",
"E",
"F",
"G",
]
df[metadata_columns] = df["run"].str.split("_").tolist()
# Create new metadata column `name` that matches the shape names in in the cell segmentation
df["name"] = df["cell_id"].apply(lambda x: f"Shape_{int(x)}")
# Add metadata column that enables spatialdata to match the measurements
# to the cell segmentation shapes (stored in sdata.shapes["cell_segmentation"]) [expects name of shapes attribute]
df["region_key"] = "cell_segmentation"
# Subset to unique cells
# TODO discuss why there are multiple measurements per cell
df = df.groupby("name").first().reset_index()
# Set all metadata columns, including the newly generated ones to the dataframe index
df = df.set_index(["run", "name", "region_key", *sorted(metadata_columns)])
df
| protein | A0A087WQ94 | A0A087WR50 | A0A087WSP5 | A0A0A6YVS2 | A0A0A6YW80 | A0A0J9YU79 | A0A0J9YUI8 | A0A0R4IZY2 | A0A0R4J083 | A0A0R4J0B4 | ... | P54726 | Q61581 | Q3V2R3 | Q6S9I0 | Q9D0J4 | Q99N93 | Q8CHY6 | P11835 | P56183 | Q6P8J7 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| run | name | region_key | A | B | C | D | E | F | G | aid | cell_id | date | method | ms | sample_id | uniprot_ref | |||||||||||||||||||||
| 20220820_TIMS06_FAS_SA_S720_scDVP_m1A_Ref10_AID8_01_S2-A1_1_4970_target4 | Shape_1 | cell_segmentation | FAS | SA | S720 | S2-A1 | 1 | 4970 | target4 | AID8 | 01 | 20220820 | scDVP | TIMS06 | m1A | Ref10 | 2546.170495 | 3112.807142 | 4247.124853 | 1906.437899 | 4825.246524 | 1589.062179 | 736.197798 | 6044.616609 | 6140.588482 | 1625.809065 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m1A_Ref10_AID8_10_S2-A10_1_4979_target4 | Shape_10 | cell_segmentation | FAS | SA | S720 | S2-A10 | 1 | 4979 | target4 | AID8 | 10 | 20220820 | scDVP | TIMS06 | m1A | Ref10 | 8077.136135 | 3330.472828 | 2268.027890 | 1526.827577 | 6940.136352 | 3275.123797 | 2150.213041 | 10486.206852 | 6641.898142 | 12554.911721 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m1A_Ref10_AID8_11_S2-A11_1_4980_target4 | Shape_11 | cell_segmentation | FAS | SA | S720 | S2-A11 | 1 | 4980 | target4 | AID8 | 11 | 20220820 | scDVP | TIMS06 | m1A | Ref10 | NaN | 4435.863428 | 5348.012649 | 1748.762739 | 10268.052605 | 1197.900908 | 2981.050155 | 12238.028228 | 8792.139128 | 1203.145811 | ... | NaN | NaN | 3487.29117 | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m1A_Ref10_AID8_12_S2-B1_1_4981_target8 | Shape_12 | cell_segmentation | FAS | SA | S720 | S2-B1 | 1 | 4981 | target8 | AID8 | 12 | 20220820 | scDVP | TIMS06 | m1A | Ref10 | 1196.769924 | 3220.892955 | 5649.794082 | 5531.072645 | 11462.061995 | 1891.362297 | 3109.722352 | 8266.772895 | 4655.184605 | 3886.250550 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m1A_Ref10_AID8_13_S2-B2_1_4982_target8 | Shape_13 | cell_segmentation | FAS | SA | S720 | S2-B2 | 1 | 4982 | target8 | AID8 | 13 | 20220820 | scDVP | TIMS06 | m1A | Ref10 | 3896.896268 | 3892.945494 | 3787.335743 | 3654.229206 | 9770.141567 | 2570.591653 | 3336.025993 | 13528.636038 | 8340.092785 | 2791.022612 | ... | NaN | 318.401047 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m3B_Ref10_AID8_75_S2-G9_1_4961_target4 | Shape_75 | cell_segmentation | FAS | SA | S720 | S2-G9 | 1 | 4961 | target4 | AID8 | 75 | 20220820 | scDVP | TIMS06 | m3B | Ref10 | 1853.740651 | 3846.307917 | 2964.671425 | 3770.539387 | 6331.494696 | 4519.779519 | 2492.729865 | 15927.070502 | 11184.127594 | 232.821475 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | 1078.283601 |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m3B_Ref10_AID8_76_S2-G10_1_4962_target4 | Shape_76 | cell_segmentation | FAS | SA | S720 | S2-G10 | 1 | 4962 | target4 | AID8 | 76 | 20220820 | scDVP | TIMS06 | m3B | Ref10 | 4479.211592 | 2225.215970 | 3729.255133 | 1500.205313 | 20813.766732 | 5020.883312 | 2230.125342 | 8551.449242 | 5701.956014 | 1810.053280 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m3B_Ref10_AID8_77_S5-G11_1_4963_target8 | Shape_77 | cell_segmentation | FAS | SA | S720 | S5-G11 | 1 | 4963 | target8 | AID8 | 77 | 20220820 | scDVP | TIMS06 | m3B | Ref10 | 3994.153689 | 2295.307398 | 2192.080927 | 15405.786639 | 1908.038186 | 3279.212194 | 2958.605514 | 7308.751211 | 4870.938330 | 1780.867609 | ... | NaN | NaN | NaN | NaN | 488.112002 | NaN | NaN | NaN | NaN | NaN |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m1A_Ref10_AID8_08_S2-A8_1_4977_target4 | Shape_8 | cell_segmentation | FAS | SA | S720 | S2-A8 | 1 | 4977 | target4 | AID8 | 08 | 20220820 | scDVP | TIMS06 | m1A | Ref10 | 3880.901353 | 3354.038617 | 3288.165942 | 3010.736507 | 5709.819890 | 3183.414270 | 2314.839162 | 16373.762603 | 11374.528812 | 3555.521124 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 20220820_TIMS06_FAS_SA_S720_scDVP_m1A_Ref10_AID8_09_S2-A9_1_4978_target8 | Shape_9 | cell_segmentation | FAS | SA | S720 | S2-A9 | 1 | 4978 | target8 | AID8 | 09 | 20220820 | scDVP | TIMS06 | m1A | Ref10 | 3086.943355 | 3421.335838 | 2606.051069 | 5119.567885 | 9104.939493 | 2330.939112 | 2840.631062 | 11393.620934 | 5431.817081 | 2077.488892 | ... | 173.200087 | 480.249257 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
77 rows × 3738 columns
# Parse dataframe
# Set anndata.AnnData.var index to protein uniprot accessors to enable querying by protein names (required for plotting)
sdata.tables["m1a_table"] = parse_df(
df, var_index="protein", region="cell_segmentation", region_key="region_key", instance_key="name"
)
# Inspect
sdata.tables["m1a_table"]
AnnData object with n_obs × n_vars = 77 × 3738
obs: 'run', 'name', 'region_key', 'A', 'B', 'C', 'D', 'E', 'F', 'G', 'aid', 'cell_id', 'date', 'method', 'ms', 'sample_id', 'uniprot_ref'
uns: 'spatialdata_attrs'
After having added the omics layer to the spatialdata object, we can overlay the proteomics measurement with the imaging. In this example, we will focus on E-Cadherin (P09803) for which the original authors also developed an antibody-based staining.
min_coordinate = np.array([4000, 8000])
size = 5000
(
sdata
# Subset to relevant area
.query.bounding_box(
axes=("x", "y"),
min_coordinate=min_coordinate,
max_coordinate=min_coordinate + size,
target_coordinate_system="global",
)
)
SpatialData object
├── Images
│ └── 'm1a_image': DataArray[cyx] (4, 5000, 5000)
├── Shapes
│ └── 'cell_segmentation': GeoDataFrame shape: (11, 3) (2D shapes)
└── Tables
└── 'm1a_table': AnnData (2, 3738)
with coordinate systems:
▸ 'global', with elements:
m1a_image (Images), cell_segmentation (Shapes)
▸ 'to_lmd', with elements:
cell_segmentation (Shapes)
sdata.tables["m1a_table"].obs["expression"] = sdata.tables["m1a_table"][:, "P09803"].X.flatten()
fig, axs = plt.subplots(1, 2, figsize=(16, 5))
min_coordinate = np.array([4000, 7000])
max_coordinate = min_coordinate + 600
(
sdata
# Subset to relevant area
.query.bounding_box(
axes=("x", "y"),
min_coordinate=min_coordinate,
max_coordinate=max_coordinate,
target_coordinate_system="global",
)
.pl.render_images("m1a_image", channel=1)
.pl.render_shapes(
element="cell_segmentation", color="P09803", norm=mpl.colors.Normalize(0, 7000), outline_alpha=1, fill_alpha=1
)
.pl.show(coordinate_systems="global", ax=axs[0])
)
axs[0].set_title("LC/MS-measurement + $\\alpha$-E-Cadherin")
(
sdata
# Subset to relevant area
.query.bounding_box(
axes=("x", "y"),
min_coordinate=min_coordinate,
max_coordinate=max_coordinate,
target_coordinate_system="global",
)
.pl.render_images("m1a_image", channel=1)
.pl.render_shapes(
element="cell_segmentation", color="P09803", outline_alpha=1, outline_color="#cccccc", fill_alpha=0
)
.pl.show(coordinate_systems="global", ax=axs[1])
)
axs[1].set_title("Cell segmentation + $\\alpha$-E-Cadherin")
Text(0.5, 1.0, 'Cell segmentation + $\\alpha$-E-Cadherin')
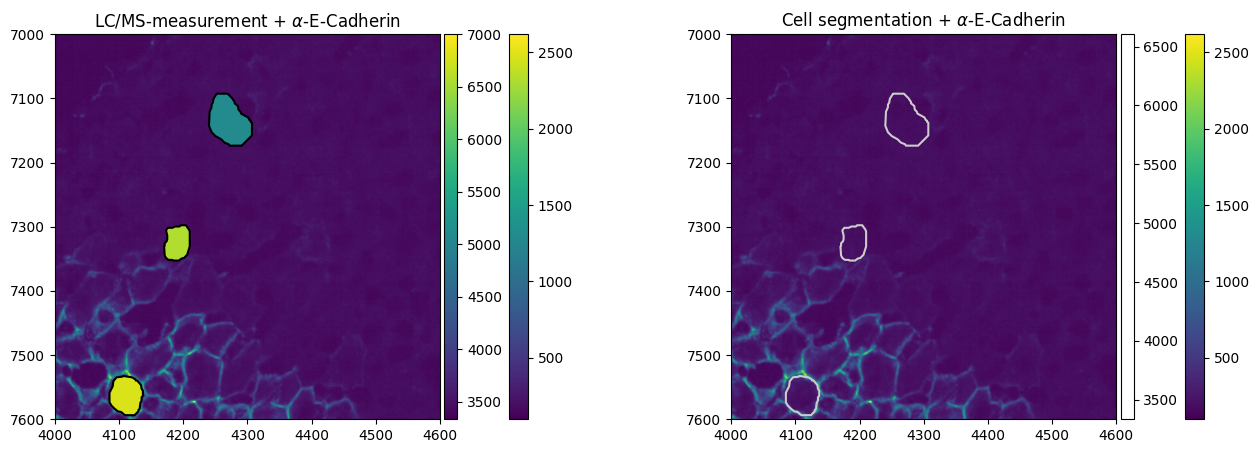
We note that the staining intensity correlates with the measured abundance of E-Cadherin in the cells.